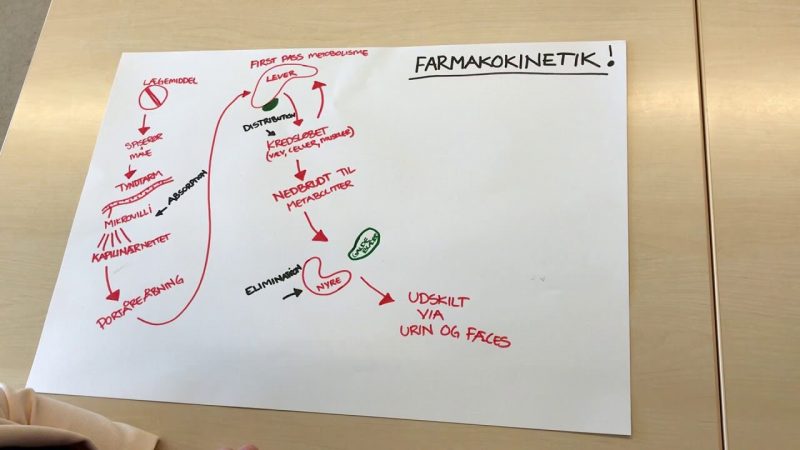
Farmakokinetik (ADME – Teori Lengkap)
Farmakokinetik adalah salah satu lingkup bahasan farmakologi yang mempelajari dan mengkarakterisasi nasib obat di dalam tubuh.
Secara umum ada empat hal yang dipelajari dalam farmakokinetik, yaitu absorbsi, distribusi, metabolisme dan ekskresi. Keempat faktor tersebut ditentukan oleh sifat fisiko kimia obat dan variasi fisiologik serta adanya penyakit, interaksi obat dan faktor lingkungan.
Adanya pemahaman farmakokinetik obat akan membantu mengupayakan pengobatan secara rasional.
DAFTAR ISI:
A. Permeasi dan Absorbsi
Absorbsi obat adalah proses pergerakan obat dari tempat obat diberikan ke dalam sirkulasi sistemik. Untuk itu semua obat harus melewati satu atau lebih membran sel agar mencapai tempat kerjanya dan untuk dapat diekskresikan tubuh.
Obat melintasi membran sel (trans membrane transport) dapat secara pasif atau aktif (energy dependent). Ada beberapa macam transport obat melintasi sel membran (trans membrane transport) atau permeasi, yaitu:
1. Difusi Pasif
Obat dapat berdifusi sesuai kelarutannya di membran sel dan gradien konsentrasinya melintasi membran. Semakin tinggi kelarutan obat dalam lemak, semakin besar melintasi membran secara difusi.
Sebagian besar obat adalah asam atau basa lemah, dan dapat berada dalam bentuk bermuatan (ionized) dan tidak bermuatan (non ionized). Jumlah relatif kedua bentuk tergantung pada pK obat dan pH dimana obat berada.
Untuk dapat melintasi membran sel diperlukan obat dalam bentuk tidak bermuatan. Besar ionisasi ditentukan oleh persamaan Henderson-Hasselbach, sebagai berikut:
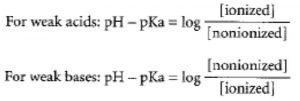
Dengan demikian, untuk obat asam (R-COOH) semakin terionisasi (R-COO) saat pH meningkat, sedangkan obat basa (R-NHZ) semakin terionisasi (R-NH3+) seiring pH menurun.
Oleh karena itu, asam lemah seperti ampisilin atau aspirin akan lebih baik diserap di pH asam seperti di lambung, sementara basa Iemah seperti amidarone akan lebih baik diserap dalam larutan yang lebih basa yaitu di usus.
Pada tabel di bawah ini akan menggambarkan besar (%) obat bentuk non ionized dalam fungsi pH lingkungan obat berada.
| pH-pKa | -2 | -1 | 0 (nol) | +1 | +2 |
| Asam lemah: % non ionized | 99 | 90 | 50 | 10 | 1 |
| Basa lemah: % non ionized | 1 | 10 | 50 | 90 | 99 |
Di samping pKa obat dan pH lingkungan, ukuran molekul juga mempengaruhi kemampuan obat melintasi membran sel. Koefisien difusi berbanding terbalik dengan akar kuadrat dari berat molekul. Jadi, untuk difusi pasif, molekul obat kecil cenderung berdifusi melintasi membran lebih mudah daripada molekul besar.
2. Difusi Difasilitasi
Difusi ion atau senyawa organik melintasi membran sel dapat difasilitasi oleh pembawa (membrane transporter). Melalui cara transport ini tidak diperlukan energi, dan senyawa akan bergerak sesuai gradien konsentrasi zat pembawa.
Pergerakan akan berhenti setelah potensial elektrokimia di kedua sisi membran mencapai keseimbangan.
3. Transportasi Aktif
Merupakan cara obat melintasi membran yang membutuhkan energi dan dan dapat bergerak melawan konsentrasi gradien, selektif, dapat diblok secara kompetitif dan dapat terjadi kejenuhan.
Proses transport ini berperan penting pada proses uptake dan efflux obat atau senyawa kimia lain, terutama terjadi di membran neuronal, pleksus koroid, hepatosit, clan tubulus ginjal.
4. Endositosis dan Eksositosis
Endositosis merupakan salah satu bentuk transport senyawa kimia yang mempunyai molekul besar. Pada endositosis, membran plasma akan menelan (engulfed) senyawa kimia (contoh vitamin B6) untuk dimasukkan dalam vesikel, kemudian dilepaskan ke sitosol dengan cara merusak vesikel tersebut.
Sebaliknya, pada eksositosis, vesikel di sitoplasma akan melakukan fusi dengan membran sel dan selanjutnya isi vesikel (contoh nor epinefrin) akan dilepaskan ke luar sel.
Secara umum, cara permeasi obat dapat dilihat pada berikut ini.
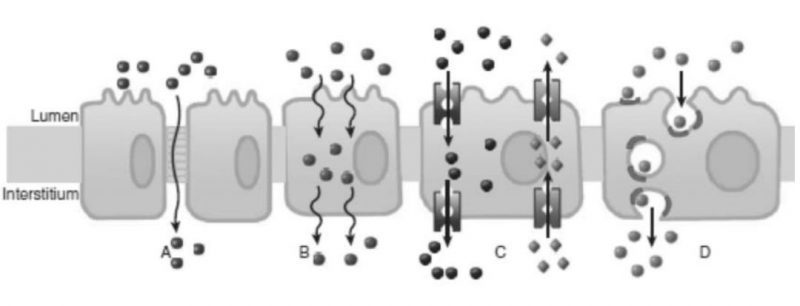
Selain proses transport melintasi membran sel, absorpsi obat juga ditentukan oleh keunikan yang terjadi di lambung dan usus. Sebagian besar proses transport obat di saluran pencernaan terjadi melalui difusi pasif, oleh karenanya penyerapan menjadi lebih baik bila obat tersebut berada dalam bentuk yang tidak bermuatan dan lebih lipofilik.
Berdasarkan konsep partisi-pH, maka obat asam lemah diprediksi akan lebih baik diserap dari lambung (pH 1 sampai 2) daripada dari usus bagian atas (pH 3 sampai 6), dan sebaliknya untuk basa lemah.
Namun, karena epitel lambung dilapisi mukus yang tebal, dan luas permukaannya sempit; sedangkan villi usus bagian atas memberikan area permukaan yang sangat luas (sekitar 200 m²), maka tingkat penyerapan obat di usus akan lebih besar dari pada lambung bahkan meskipun obat tersebut sebagian besar terionisasi di usus dan sebagian besar tidak bermuatan di lambung.
Dengan demikian, setiap faktor yang mempercepat pengosongan lambung akan cenderung meningkatkan tingkat penyerapan obat, sementara faktor apa pun yang menunda pengosongan lambung akan memiliki efek sebaliknya, terlepas dari karakteristik obatnya.
Difusi pasif melalui interseluler (A) atau plasmalemma (B), Transport aktif menggunakan protein carriers dari atau ke dalam sel (C), dan endositosis dan eksositosis (Katzung, et al., 2015).
Faktor lain dapat mempengaruhi penyerapan obat adalah laju aliran darah, jumlah cairan di lokasi penyerapan dan bentuk sediaan obat. Tingkat disolusi merupakan faktor pembatas dalam penyerapan obat bentuk padat, terutama jika obat tersebut memiliki kelarutan dalam air yang rendah atau jumlah cairan di lokasi penyerapan minimal.
Kecepatan dan besar obat yang diabsorpsi dapat membawa dampak pada efektivitas obat secara klinis. Penggunaan obat pada dosis tunggal, obat yang lebih cepat dan lebih banyak yang diabsorpsi akan mempunyai mula dan durasi obat yang lebih baik.
Tetapi, pada penggunaan obat berulang, adanya perbedaan kecepatan dan besar absorbsi antar obat tidak menunjukkan perbedaan rata-rata konsentrasi obat dalam plasma.
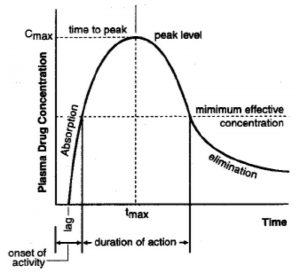
- Cmax: konsentrasi maksimal obat dalam plasma;
- tmax: waktu yang diperlukan obat mencapai Cmax;
- AUC; area under curve yang menunjukkan jumlah obat dalam plasma (Kaplan).
B. Bioavailabilitas dan First Pass Metabolism
Bioavailabilitas didefinisikan sebagai fraksi obat yang diberikan yang mencapai sirkulasi sistemik dalam bentuk yang tidak berubah.
Bioavailabilitas selain ditentukan oleh jumlah absorpsi obat, juga dipengaruhi besar metabolisme dan /atau ekskresi yang mungkin terjadi sebelum obat tersebut mencapai sirkulasi sistemik. Pada gambar sebelumnya di atas terlihat bahwa besar bioavailabilitas sebanding dengan luas area di bawah kurva (are under the blood concentration-time curve).
Karena pemberian obat secara intravena secara langsung menyimpan obat ke dalam sirkulasi sistemik, bioavailabilitas obat yang diberikan lV didefinisikan sebagai 100%, sedangkan dengan rute lain kurang dari 100% akibat penyerapan yang tidak lengkap atau metabolisme sebelum obat mencapai sirkulasi sistemik.
Pada pemberian per oral, setelah terjadi penyerapan obat di seluruh dinding usus, obat didistribusikan ke liver terlebih dahulu melalui sirkulasi portal sebelum masuk sirkulasi sistemik. Seperti yang diketahui bahwa liver adalah tempat utama metabolisme obat, maka sejumlah obat akan mengalami in aktivasi karena metabolisme sebelum mencapai sirkulasi sistemik.
Fenomena ini disebut sebagai first pass metabolism (metabolisme lintas pertama) yang merupakan faktor utama penentu besar bioavailabllitas obat. Efek dari metabolisme lintas pertama dinyatakan sebagai extraction ratio, yang besarnya dapat ditentukan dengan rumus:
ER = CLliver/Q
Q adalah aliran darah ke liver yang biasanya sebesar 90 L/jam untuk orang normal dengan berat badan 70 kg, Cl liver merupakan klirens liver.
Selanjutnya besar biovaibilitas (F) ditentukan dengan rumus:
F = f x (1 – ER)
f adalah besar absorpsi obat, ER adalah besar extraction ratio
Contoh: Morfin adalah obat yang dapat diabsorpsi secara sempurna. Klirens hepatik morfm sebesar 60L/70 kg sedangkan aliran darah heoatik 90L/70 kg atau extraction retaio morfin adalah 0.67.
Dengan demikian meskipun morfin dapat diabsorpsi sempurna tetapi bioavailabilitasnya hanya F = 1 – ER atau 33%.
C. Rute Pemberian Obat dan Bioavailabilitas
Ada dua kategori umum rute pemberian obat yaitu enteral, yang memanfaatkan saluran pencernaan, dan parenteral, yang tidak. Lihat pembahasan sub bab ini selengkapnya di sini.
D. Distribusi
Obat yang berada di dalam sirkulasi sistemik akan segera diditribusikan ke organ dan jaringan. Proses tersebut melibatkan permeasi membran sel, yang dipengaruhi kelarutan obat di dalam lemak (bentuk tidak bermuatan), kecepatan aliran darah menuju jaringan dan ikatan molekul obat dengan protein plasma.
Meskipun obat terlarut dalam cairan plasma, namun sebagian besar obat berikatan dengan komponen plasma antara lain albumin, globulin, transferrin, ceruloplasmin, glycoprotein, α1 dan β- lipoprotein. Obat asam pada umumnya terikat pada albumin, sedangkan obat basa pada lipoprotein dan α1-acid glycoprotein.
Besar obat yang terikat protein plasma mempengaruhi distribusi obat dan kecepatan eliminasi karena hanya obat bebas yang dapat melintasi membran sel untuk dapat mencapai target obat, mengalami metabolisme dan ekskresi.
Ikatan obat dengan protein plasma (albumin atau α1-acid glycoprotein) bersifat non selektif, oleh karena itu obat yang mempunyai sifat fisikokimia serupa dengan obat lain dapat saling bersaing untuk berikatan dengan plasma protein yang sama (albumin atau α1-acid glycoprotein).
Keadaan ini menyebabkan obat yang mempunyai afinitas ikatan dengan protein plasma yang rendah menjadi bebas dan dapat terdistribusi ke target organ sehingga efek farmakologik obat meningkat.
Kesamaan fisikokimia juga bisa terjadi antara obat dan zat endogen (misal sulfonamide dan bilirubin), yang menyebabkan adanya bilirubin yang tidak terkonjugasi tidak terikat albumin sehingga timbul risiko ensefalopati bilirubin pada bayi baru lahir.
Obat di dalam tubuh terdistribusi secara merata, beberapa jaringan/organ dapat menjadi tempat terakumulasinya obat-obat tertentu, antara lain:
- Ginjal mengandung protein metallothionein yang mempunyai afinitas tinggi terhadap kadmium, merkuri dan timah sehingga konsentrasi logan tersebut tinggi di ginjal.
- Traktus uveal sering menjadi tempat simpanan klorpromazin dan fenotiazin sehingga efek samping dari keduanya salah satunya adalah kerusakan retina.
- Obat-obat yang sangat larut lemak akan tersimpan banyak pada lemak tubuh.
- Obat-obat yang mempunyai kelarutan lemak tinggi dan pKA lebih dari 8 (contoh antihistamin, imipramin, amfetamin, metadon, klorpromazin) dapat terakumulasi di paru.
- Obat-obat tertentu (contoh tetrasiklin, timah, cisplastin) dapat diserap ke dalam permukaan kristal tulang.
Di dalam tubuh ada beberapa barier khusus yaitu sawar otak (blood brain barrier). Membran kapiler antara plasma dan otak berbeda dengan tempat lain (plasma dengan organ lain) dimana sel endotel pada sawar otak mempunyai ikatan yang sangat ketat sehingga untuk melewatinya obat harus melalui sel bukan antar sel.
Hal tersebut menyebabkan hanya obat yang mempunyai koefisien partisi |ipid:air yang tinggi yang dapat menembus sawar otak. Disamping itu karena cairan serebrospinal mempunyai pH 7,35 maka hanya obat basa yang dapat terkonsentrasi di otak, sedangkan obat asam akan dikeluarkan.
E. Metabolisme
Metabolisme, redistribusi dan ekskresi merupakan proses yang menentukan masa kerja obat dan kecepatan eliminasi obat. Pada proses metabolisme sebagian obat diubah menjadi metabolit yang kurang mempunyai aktivitas farmakologik dan menjadi lebih polar (larut air) sehingga mudah untuk diekskresikan.
Metabolisme terjadi terutama di hepar, yang melibatkan enzim oksidasi dan reduksi (fase I) dan enzim konjugasi (fase ll).
Pada metabolisme obat fase I, sebagian besar obat mengalami oksidasi oleh enzim cytochrome P450 (CYP 450) super family. Di antara famili CYP 450, CYP3A4 merupakan isoform enzim yang dominan baik dalam jumlah yang ada di liver (50%) maupun variasi obat yang merupakan substrat enzim tersebut.
Selanjutnya isoform enzim CYP 450 terbanyak lainnya yang terlibat dalam metabolisme obat adalah CYP2D6 (30%), dan CYP2C9 (10%).
Daftar obat penghambat dan penginduksi enzim metabolisme di liver| Penghambat | Penginduksi |
|---|---|
| Kloramfenikol | Barbiturat |
| Simetidin | Karbamazepin |
| Siprofloksasin | Deksametazon |
| Klaritomisin | Fenitoin |
| Eritronisin | Rifampisin |
| Flukonazol | |
| Isoniazid (INH) | |
| Ketonazol | |
| Ritonavir | |
| Catatan: jus jeruk | Catatan: konsumsi alkohol kronik, asap rokok. |
Cytochrome P450 merupakan enzim yang dapat berubah aktivitasnya menjadi lebih tinggi atau rendah akibat adanya obat lain atau penyakit. Beberapa obat dapat menghambat metabolisme obat lain, dikarenakan obat obat tersebut mempunyai ikatan dengan enzim pada tempat aktif yang sama atau enzim mengalami inaktivasi (suicidal inactivation).
Fenomena hambatan metabolisme obat merupakan keadaan yang paling sering terjadi terkait modulasi CYP 450 dan interaksi antar obat. Di sisi lain aktivitas enzim CYP 450 juga dapat distimulasi/induksi oleh obat lain atau obat yang sama melalui peningkatan pembentukan enzim baru atau penurunan degradasi enzim proteolitik.
Adanya hambatan enzim potensial menimbulkan toksisitas, sedangkan adanya induksi enzim memungkinan terjadi kegagalan terapi karena obat tidak mencapai konsentrasi yang semestinya. Pada tabel di atas terlihat beberapa contoh obat yang dapat menghambat atau menginduksi enzim metabolisme di liver.
Pada metabolisme obat fase II, obat akan mengalami konjugasi sehingga meningkatkan sifat kelarutan obat di dalam air dan menjadikan ekskresi obat lebih mudah. Fase ini dapat terjadi setelah obat mengalami metabolisme fase sebelumnya atau langsung tanpa melalui metabolisme obat fase I.
Enzim konjugasi yang terlibat pada metabolisme obat antara lain Glucuronosyl transferases, N acetyltransferase, Sulfotransferases, dan methyltransferases. Enzim enzim tersebut mempunyai banyak isoform. Seluruh enzim metabolisme tersebut selain berada di liver, juga ditemukan di dinding lambung, ginjal, otak, plasenta, kulit dan paru.
Dalam metabolisme, beberapa obat justru dikonversi menjadi bentuk molekul yang mempunyai efek farmakologi lebih baik atau menjadi metabolit yang toksik. Prodrugs adalah senyawa yang tidak aktif secara biologis yang dirancang untuk memaksimalkan jumlah senyawa aktif yang mencapai tempat kerjanya.
Sebagian besar prodrugs dikonversi dengan cepat menjadi metabolit aktif melalui hidrolisis ikatan ester atau amida. Metabolit aktif yang terbentuk dapat memiliki:
- aktivitas yang secara kualitatif mirip dengan obat induk tapi secara kuantitatif berbeda,
- aktivitas yang berbeda secara kualitatif,
- toksisitas lebih besar,
- masa paruh yang berbeda.
Contoh prodrugs antara lain diazepam (Valium), yang didemetilasi untuk membentuk metabolit dengan waktu paruh lebih lama namun efek farmakologisnya lebih rendah. Acetaminophen merupakan salah satu contoh obat yang menghasilkan metabolit toksik Nasetilpbenzoquinon imine (NAPQl), yang merupakan hasil glukuronidasi dan sulfasi.
Overdosis acetaminophen dapat menyebabkan penipisan tingkat GSH seluler, sehingga meningkatkan potensi NAPQI untuk berinteraksi dengan komponen seluler lainnya. Toksisitas asetaminofen dikaitkan dengan peningkatan kadar NAPQl dan nekrosis jaringan terutama liver.
F. Ekskresi
Ekskresi merupakan proses berpindahnya obat dari lingkungandalamkeluar tubuh. Ginjal merupakan organ utama yang berperan pada ekskresi obat. Besar ekskresi obat merupakan hasil resultante antara tiga proses yaitu filtrasi glomerulus, sekresi aktif, dan reabsorbsi pasif.
Filtrasi glomerulus merupakan fungsi linier yang tidak jenuh, dapat memfilter obat yang bermuatan atau tidak bermuatan tetapi tidak dapat memfilter obat yang terikat protein plasma atau yang mempunyai berat molekul yang relatif besar.
Dengan demikian faktor yang mempengaruhi ikatan obat dengan protein plasma akan mengubah filtrasi obat yang selanjutnya mempengaruhi waktu paruh obat (waktu yang dibutuhkan untuk obat menjadi separuh konsentrasi).
Sekresi aktif yang terjadi di tubulus proksimal memainkan peran kecil dalam ekskresi fisiologis normal, namun berperan penting dalam ekskresi beberapa obat. Sistem sekresi ini dapat memfasilitasi obat-obat yang tidak dapat melalui filtrasi glomerulus karena obat yang bermuatan baik anion atau kation sering berikatan dengan protein plasma.
Namun karena ikatan dengan protein bersifat reversibel, maka sistem sekresi aktif dapat secara cepat dan efisien memindahkan obat yang terikat protein dari darah ke tubulus. Di dalam sistem sekresi tubular aktif, obat dapat menjadi substrat untuk dua sistem, yaitu sekresi untuk organik anion dan organik kation.
Antara kedua substrat obat tersebut dapat saling berkompetisi sehingga salah satu obat dapat mengalami penghambatan sekresi. Obat yang mengalami filtrasi dapat direabsorpsi di sel tubulus ginjal. Transportasi aktif di tubulus distal dikaitkan secara fisiologis dengan asam urat endogen, glukosa dan asam amino.
Difusi nonionik pasif terjadi di seluruh lumen tubular kembali ke sirkulasi sistemik, yang tergantung pada gradien konsentrasi, gradien pH, kelarutan lipid, derajat ionisasi, dan ukuran molekul.
Peningkatan keasaman urin tubular sangat mempengaruhi laju reabsorpsi elektrolit lemah. Bentuk yang tidak terion dapat segera menyebar kembali ke sirkulasi, sedangkan bentuk terionisasi terjebak dalam urin di tubulus dan diekskresikan. Sebaliknya dengan urin alkalin membantu reabsorpsi basa lemah dan ekskresi asam lemah.
Dalam overdosis obat, manipulasi pH urin kadang digunakan untuk mencegah reabsorpsi. Pemberian ammonium chloride menghasilkan urine asam sedangkan sodium bikarbonat menghasilkan urin alkali.
Dengan demikian memanipulasi pH urin merupakan salah satu cara menangani keracunan obat karena dapat meningkatkan ekskresi obat. Pada gambar ekskresi obat melalui ginjal di bawah ini terlihat proses filtrasi glomerulus, sekresi aktif, dan reabsorpsi tubular.
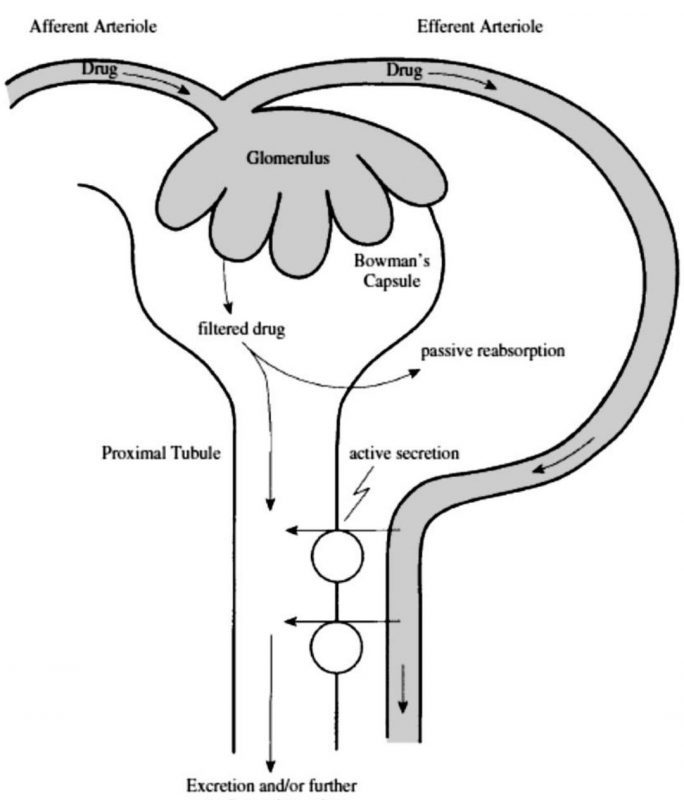
Filtrasi terjadi pada obat yang tidak terikat protein plasma melalui pori kapiler glomerulus. Obat yang bersifat larut lemak dapat mengalami reabsorpsi melalui nefron. Sekresi aktif asam organik dan basa organik terjadi hanya di tubulus proksimal (Craig, Modern Pharmacology).
G. Sirkulasi Enterohepatik
Sirkulasi enterohepatik merupakan siklus obat dari hati empedu usus portal vena hati. Siklus ini penting untuk obat atau metabolit yang terkonjugasi karena obat/ metabolit terkonjugasi bersifat sangat polar dan mempunyai berat moleukul yang tinggi.
Metabolisme terkonjugasi yang disekresikan dalam usus akan mengalami hidrolisis oleh enzim usus (contoh glukoronidase) menghasilkan obat asli, yang lebih mudah larut dalam lemak dan tidak terion, dan karena itu siap mengalami reabsorpsi.
Hal ini penting secara farmakokinetik untuk obat tertentu (digitoxim morfin) karena dapat memperpanjang waktu paruh biologis atau obat lain (kloramfenikol) menyebabkan toksisitas. Beberapa obat yang mengalami siklus enterohepatik adalah estradiol, metronidazol, fenitoin, morfm dan indometasin.
Selain melalui ginjal, obat juga dapat diekskresikan melalui cairan tubuh lain yaitu antara lain saliva, keringat, dan air susu ibu. Obat yang diekskresikan melalui saliva atau keringat tergantung pada obat yang larut lemak dan dapat melalui sel epitel glandula keduanya.
Dengan demikian berapa banyak obat yang diekskresikan malalui saliva atau keringat sangat tergantung pada pKa obat dan pH individu.
Pada air susu ibu menyusui ditemukan obat, yang jumlahnya tergantung pada kadar obat plasma ibu, kelarutan obat dalam lemak, dan besar ionisasi. Dengan demikian jumlah dalam air susu ibu (ph 6,5) untuk obat basa akan lebih banyak dibandingkan obat asam.
Untuk obat yang sangat larut lemak akan berikatan dengan lemak susu sedangkan obat yang larut air akan lebih banyak berada di air susu. Dalam meminimalisasi paparan obat pada bayi, sebaiknya ibu menggunakan obat sesingkat mungkin dan minum obat setelah menyusui.
Beberapa obat yang dieksdresikan melalui air susu ibu antara lain asam salisilat, barbiturat, kafein, narkotik, dan alkohol.
H. Variasi dalam Farmakokinetik Obat
Terapi akan jauh lebih mudah jika respons tubuh sama terhadap dosis obat yang sama. Kenyataannya variasi interpersonal dan intraindividual seringkali besar sehingga konsentrasi obat pada target obat berbeda satu dengan yang lain.
Variasi farmakokinetik dapat terkait absorpsi, distribusi, metabolisme atau ekskresi obat, yaitu antara lain:
1. Usia
Perhatian yang khusus harus dilakukan dalam memberikan obat apapun kepada neonatus, bayi, dan orang tua. Pada bayi kecepatan pengosongan lambung sesuai dengan umur dan formulasi makanan yang diasup.
Pada bayi berumur sampai dengan satu tahun, pengosongan lambung dan peristataltik usus cenderung lambat, tetapi pada umur 2-12 tahun kecepatan pengosongan menjadi semakin lebih cepat sehingga absorpsi obat juga lebih cepat.
Total cairan tubuh janin, bayi baru lahir atau bayi lebih banyak dibandingkan anak-anak atau dewasa. Keadaan ini menyebabkan besar volume distribusi untuk obat yang larut air akan meningkat sehingga untuk beberapa obat dibutuhkan pemberian loading dose.
Sebaliknya dengan obat yang larut lemak, karena bayi mempunyai lemak tubuh yang sedikit maka obat yang tersimpan di lemak juga sedikit. Pada bayi fungsi hati dan ginjal belum sempurna sehingga dapat timbul toksisitas pada penggunaan obat yang diberikan berulang.
Contoh: pemberian antibiotik kloramfenikol (biasanya terkonjugasi dalam hati dan diekskresikan dalam urin) kepada bayi yang baru lahir menyebabkan toksisitas dan kematian serius (“sindrom bayi abu-abu”).
Konjugasi lambat juga merupakan salah satu alasan mengapa morfin (yang diekskresikan terutama sebagai glukuronida) tidak digunakan sebagai analgesik dalam persalinan, karena obat yang ditransfer melalui plasenta memiliki waktu paruh yang lama pada bayi yang baru lahir dan dapat menyebabkan depresi pernafasan berkepanjangan.
Pada orang tua, proses absorpsi tidak lengkap, pengosongan lambung dan produksi asam lambung menurun serta terjadi hipoalbumin. Begitu juga dengan fungsi ginjal yang memperlihatkan tingkat filtrasi glomerulus yang menurun perlahan dari sekitar 20 tahun, turun sekitar 25% pada 50 tahun dan 50% pada 75 tahun.
Pada fungsi metabolime, aktivitas enzim mikrosomik hati menurun perlahan (dan sangat bervariasi) seiring bertambahnya usia, sedangkan volume distribusi obat terlarut lipid meningkat, karena proporsi tubuh yang bertambah dengan bertambahnya usia lanjut.
Untuk mencegah timbulnya toksisitas obat, maka pemberian obat pada orang tua sebaiknya tepat obat, tepat dosis dan selalu dilakukan monitoring kemungkinan timbulnya efek samping obat.
2. Kehamilan
Pada kehamilan terjadi perubahan fisiologis yang dapat mempengaruhi disposisi obat pada ibu dan janin. Konsentrasi albumin plasma ibu yang berkurang, dapat mempengaruhi pengikatan obat.
Sedangkan curah jantung yang meningkat, menyebabkan peningkatan aliran darah ginjal dan GFR sehingga eliminasi obat juga meningkat. Obat yang bersifat larut lemak dengan cepat dapat melintasi barier plasenta, selanjutnya mengalami metabolisme dan eksresi.
Tetapi ginjal janin bukanlah jalur eliminasi yang efisien karena obat yang diekskresikan memasuki cairan amnion dapat ditelan oleh janin.
3. Polimorfisme Genetika Farmakogenetik
Perbedaan tingkat ekspresi enzim metabolik tertentu atau bentuk enzim yang tidak normal dapat menyebabkan perbedaan individual dalam tingkat metabolisme obat di antara populasi pasien.
Contoh klasik terlihat untuk asetilasi isoniazid yang disebut “asetilator lambat“ memiliki tingkat enzim yang berkurang dan dengan demikian memiliki kadar isoniazid plasma lebih tinggi dibandingkan dengan “normal.”
Fenotip asetilator yang lambat diwarisi sebagai sifat resesif autosomal, dan terlihat pada sekitar 50% orang kulit hitam dan kulit putih di Amerika Serikat.
Polimorfisme genetik karena allel yang berbeda dari isoform yang diberikan juga terjadi. CYP2D6 memetabolisme banyak obat yang biasa diresepkan termasuk Hecanide antiaritmia, metoprolol b-blocker, dan antidepresan fiuoxetine dan imipramine.
Pemeriksaan rasio debrisoquin/4-hydroxydebrisoquin dalam urin adalah cara mudah untuk menentukan aktivitas CYP2D6.
4. Kondisi Patologis
i. Penyakit Ginjal
Penurunan fungsi ginjal dapat mempengaruhi farmakokinetik obat yang normal. Hal ini dapat menyebabkan toksisitas (digoksin) atau ketidakefektifan (vitamin D).
Hanya sedikit obat yang menimbulkan masalah serius namun masalah mungkin timbul bila kondisi berikut ada:
- Fungsi ginjal di bawah 50% normal;
- Ekskresi urin obat yang tidak berubah atau metabolit aktif > 50%;
- Obat memiliki indeks terapeutik rendah.
Obat yang menimbulkan masalah potensial pada penyakit ginjal meliputi: aminoglikosida (gentamisin, kanamisin, streptomisin); Cephaloridine; Kolistin; Digoxin; Etambutol; Lithium; Metotreksat; Procainamide; Sulfonamida.
ii. Penyakit Hati
Penurunan fungsi hati dapat mengubah farmakokinetik obat jika metabolisme hati atau ekskresi empedu merupakan rute eliminasi utama.
Daftar Pustaka
- Craig CR, Stitzel RE. 2004. Modern Pharmacology with Clinical Applications. USA: Lippincott William and Wilkins.
- Brunton L, Chabner B, Goodman LS, Knollman B. 2011. Goodman and Gilman’s Pharmacological Basis of Therapeutics. 12th edition. USA: McGraw-Hill Companies.
- Katzung BG, Trevor Aj. 2015. Basic and Clinical Pharmacology 13th. edition. USA: McGraw Hill Companies.
- Ritter J, Flower R, Henderson G, Rang H. 2015. Rang and Dale’s Pharmacology. 8th edition. UK: Churchill Livingstone.
- USMLE. 2017. Step 1 Lecture Notes Pharmacology 2017. USA: Kaplan Medical.