Thalasemia

Disclaimer Medis: Informasi pada halaman ini bersifat edukatif dan tidak menggantikan saran, diagnosis, atau pengobatan dari tenaga medis profesional. Selalu konsultasikan kondisi kesehatan Anda dengan dokter atau apoteker yang kompeten.
Apa Itu Thalasemia?
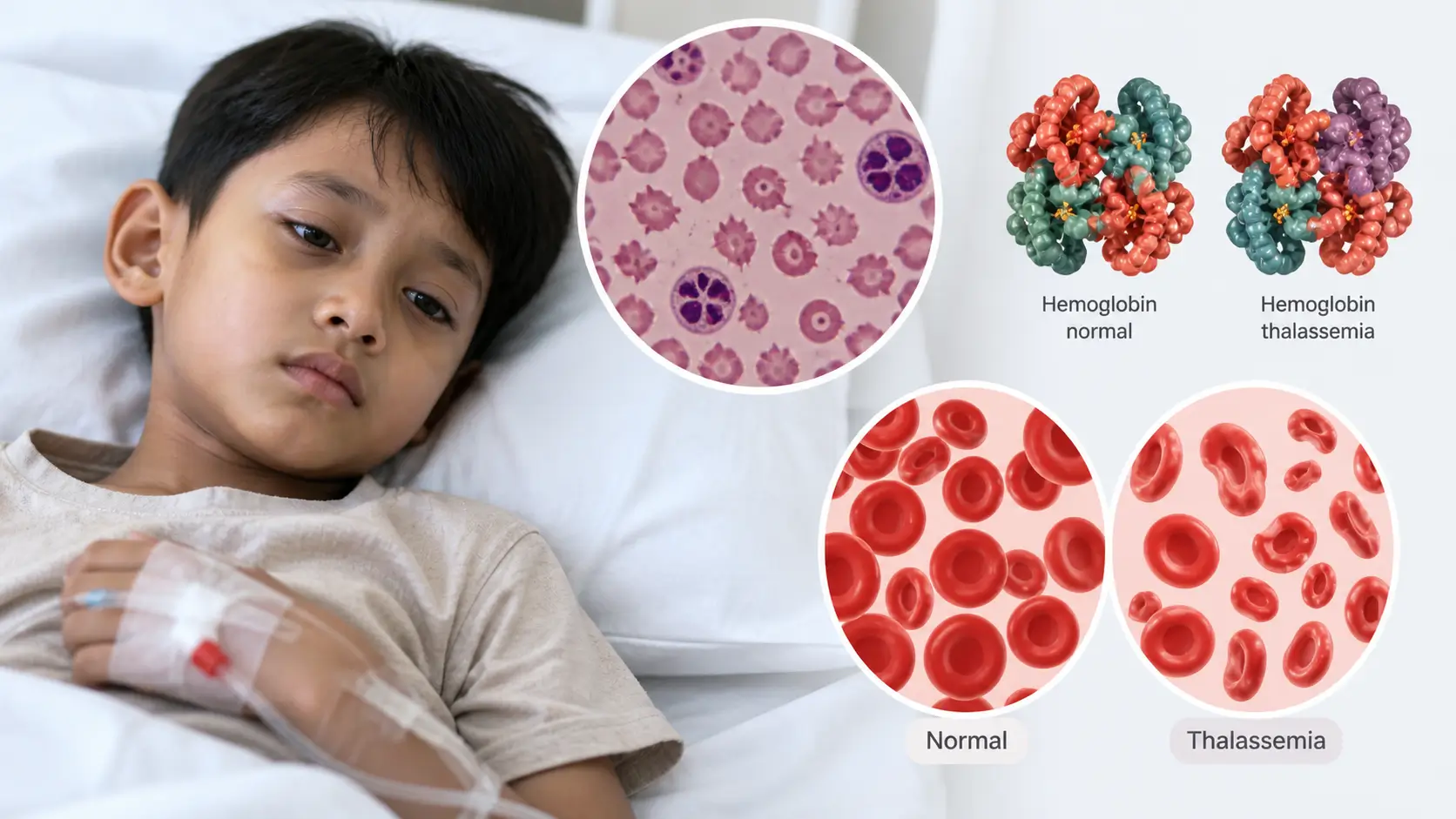
Penyakit Thalasemia adalah sekelompok kelainan darah herediter (keturunan) yang ditandai oleh berkurangnya atau tidak adanya produksi hemoglobin normal — protein dalam sel darah merah yang bertugas membawa oksigen ke seluruh tubuh. Kondisi ini terjadi akibat mutasi pada gen yang mengatur pembentukan rantai globin, yakni komponen utama hemoglobin.
Secara normal, hemoglobin dewasa (HbA) terdiri dari dua rantai alfa dan dua rantai beta. Pada thalasemia, salah satu atau kedua jenis rantai ini diproduksi dalam jumlah yang tidak cukup, sehingga sel darah merah menjadi rapuh, mudah pecah, dan berumur pendek. Akibatnya, tubuh kekurangan sel darah merah yang sehat, menyebabkan kondisi yang dikenal sebagai anemia.
Thalasemia dibagi menjadi dua tipe utama berdasarkan rantai yang terdampak: Thalasemia Alfa (mutasi gen HBA1/HBA2 pada kromosom 16) dan Thalasemia Beta (mutasi gen HBB pada kromosom 11). Masing-masing tipe memiliki spektrum keparahan yang bervariasi — dari kondisi pembawa tanpa gejala (trait/minor) hingga bentuk berat yang mengancam jiwa (mayor/intermedia).
Thalasemia merupakan penyakit genetik autosomal resesif, artinya seseorang harus mewarisi gen bermutasi dari kedua orang tua untuk mengalami bentuk yang berat. Jika hanya mewarisi dari satu orang tua, ia disebut 'pembawa' (carrier) dan umumnya tidak bergejala atau hanya bergejala ringan.
Indonesia termasuk negara dalam 'sabuk thalasemia' dunia — wilayah dengan prevalensi tinggi yang membentang dari Afrika Utara, Mediterania, Timur Tengah, Asia Selatan, hingga Asia Tenggara. Diperkirakan 3–8% penduduk Indonesia adalah pembawa gen thalasemia, menjadikannya salah satu masalah kesehatan genetik terbesar di tanah air.
Gejala Thalasemia
- Pucat atau warna kulit yang terlihat lebih putih dari biasanya, terutama terlihat pada telapak tangan, kuku, dan selaput dalam kelopak mata, yang merupakan tanda klasik anemia.
- Rasa lelah dan lemah yang berlebihan bahkan setelah istirahat cukup, karena sel darah merah tidak mampu mengantarkan oksigen yang cukup ke otot dan jaringan tubuh.
- Sesak napas atau napas yang terasa berat saat beraktivitas ringan sekalipun, akibat tubuh bekerja keras mengkompensasi kekurangan oksigen dalam darah.
- Warna kulit kekuningan (jaundice/ikterus) dan mata yang tampak kuning, disebabkan oleh pemecahan sel darah merah yang berlebihan sehingga kadar bilirubin meningkat.
- Perut membesar dan terasa penuh, terutama akibat pembesaran limpa (splenomegali) dan hati (hepatomegali) yang terjadi karena kedua organ ini bekerja ekstra untuk menghancurkan sel darah merah yang rusak.
- Wajah dan tulang mengalami perubahan bentuk pada kasus berat, seperti tulang pipi yang menonjol, rahang membesar, dan dahi yang lebih lebar, akibat sumsum tulang yang terus berekspansi mencoba memproduksi lebih banyak sel darah merah.
- Pertumbuhan yang terlambat pada anak-anak, ditandai dengan tinggi badan dan berat badan yang berada di bawah kurva pertumbuhan normal untuk usianya.
- Urine berwarna lebih gelap dari normal (coklat atau kemerahan) akibat peningkatan produk pemecahan hemoglobin yang dikeluarkan melalui ginjal.
- Denyut jantung yang cepat atau tidak teratur (palpitasi), sebagai respons kompensasi jantung terhadap rendahnya kadar oksigen dalam darah.
- Pada bayi dan anak kecil dengan thalasemia beta mayor, gejala biasanya mulai muncul antara usia 6 bulan hingga 2 tahun, berupa pucat progresif, rewel, dan sulit makan.
Penyebab Thalasemia
Penyakit Thalasemia disebabkan oleh mutasi atau delesi (kehilangan) pada gen yang mengkode rantai globin pembentuk hemoglobin, dan selalu diturunkan dari orang tua kepada anak.
Thalasemia Alfa disebabkan oleh delesi atau mutasi pada satu hingga empat gen HBA1 dan HBA2 yang terletak pada kromosom 16. Makin banyak gen yang terdampak, makin berat kondisinya — mulai dari pembawa diam (1 gen), trait alfa (2 gen), penyakit HbH (3 gen), hingga Hydrops Fetalis yang fatal (4 gen).
Thalasemia Beta disebabkan oleh mutasi titik (point mutation) pada gen HBB di kromosom 11 yang menyebabkan rantai beta globin diproduksi dalam jumlah sangat sedikit (beta+) atau sama sekali tidak diproduksi (beta0). Lebih dari 200 varian mutasi telah diidentifikasi di seluruh dunia.
Mekanisme kunci kerusakan sel darah merah: ketidakseimbangan produksi rantai globin menyebabkan rantai yang kelebihan mengendap di dalam sel, memicu oksidasi, kerusakan membran eritrosit, dan penghancuran dini sel darah merah dalam sumsum tulang maupun limpa (eritropoiesis inefektif dan hemolisis).
Penurunan bersifat autosomal resesif: kedua orang tua harus membawa setidaknya satu salinan gen bermutasi. Jika kedua orang tua adalah pembawa (carrier), setiap kehamilan memiliki peluang 25% anak lahir dengan thalasemia berat, 50% menjadi pembawa, dan 25% normal.
Faktor Risiko
- Memiliki orang tua yang keduanya adalah pembawa (carrier) gen thalasemia merupakan faktor risiko paling utama dan menentukan, karena penurunan thalasemia bergantung sepenuhnya pada warisan genetik dari kedua orang tua.
- Berasal dari atau memiliki leluhur dari wilayah endemis thalasemia, meliputi wilayah Mediterania (Italia, Yunani, Siprus), Timur Tengah, Afrika Sub-Sahara, Asia Selatan (India, Pakistan, Bangladesh), dan Asia Tenggara termasuk Indonesia, Malaysia, Thailand, dan Filipina.
- Perkawinan antar kerabat (konsanguinitas) meningkatkan risiko keduanya membawa mutasi gen yang sama, sehingga meningkatkan peluang anak yang lahir menderita thalasemia berat.
- Riwayat keluarga thalasemia — memiliki saudara kandung, paman, bibi, atau kerabat dekat yang telah terdiagnosis thalasemia, baik mayor maupun minor, merupakan sinyal penting untuk dilakukannya skrining genetik.
- Etnis tertentu di Indonesia memiliki prevalensi pembawa yang lebih tinggi, termasuk populasi di Sumatera, Kalimantan, Sulawesi Tengah, dan beberapa wilayah di Jawa, meskipun thalasemia dapat ditemukan di seluruh kelompok etnis Indonesia.
Komplikasi yang Perlu Diwaspadai
Kelebihan zat besi (iron overload/hemosiderosis) adalah komplikasi paling umum dan berbahaya pada penderita thalasemia yang rutin mendapat transfusi darah, karena setiap kantong darah membawa zat besi tambahan yang tidak dapat dikeluarkan tubuh secara alami.
Kerusakan jantung akibat penumpukan besi di otot jantung (kardiomiopati) dapat menyebabkan gagal jantung kongestif, aritmia, dan menjadi penyebab kematian utama pada penderita thalasemia yang tidak mendapat terapi kelasi besi yang adekuat.
Kerusakan hati (sirosis hepatis) dan pankreas (diabetes melitus) yang disebabkan oleh deposit besi berlebihan pada organ-organ vital tersebut, mengganggu fungsinya secara permanen.
Pembesaran limpa progresif (hipersplenisme) yang dapat menyebabkan limpa menghancurkan sel darah merah terlalu cepat, sehingga transfusi menjadi kurang efektif dan memerlukan tindakan pengangkatan limpa (splenektomi).
Osteoporosis dan deformitas tulang akibat ekspansi sumsum tulang yang berlebihan, meningkatkan risiko patah tulang dan nyeri tulang kronis.
Gangguan pertumbuhan dan perkembangan pada anak, termasuk pubertas terlambat, perawakan pendek, dan keterlambatan perkembangan seksual, akibat anemia kronis dan kelebihan besi yang memengaruhi produksi hormon.
Infeksi berulang dan berat, terutama setelah splenektomi, karena limpa berperan penting dalam sistem imun tubuh untuk melawan bakteri tertentu seperti Streptococcus pneumoniae.
Batu empedu (cholelithiasis) akibat pemecahan hemoglobin yang berlebihan menghasilkan banyak bilirubin yang dapat mengendap menjadi batu di kantung empedu.
Kapan Harus ke Dokter?
- Segera bawa anak ke dokter atau UGD rumah sakit jika tampak sangat pucat secara tiba-tiba, napas cepat dan dangkal, atau tampak sangat lemas — ini bisa menandakan kadar hemoglobin yang turun drastis (krisis aplastik atau hemolitik).
- Konsultasikan ke dokter jika bayi atau anak Anda mengalami pucat progresif, kulit dan mata menguning, perut membesar, pertumbuhan lambat, atau sering rewel tanpa sebab jelas, terutama jika salah satu atau kedua orang tua memiliki riwayat thalasemia atau anemia.
- Pasangan yang berencana menikah atau sedang merencanakan kehamilan sangat dianjurkan untuk melakukan skrining thalasemia lebih dulu, khususnya jika berasal dari daerah atau keluarga dengan riwayat thalasemia.
- Penderita thalasemia yang sedang dalam program transfusi harus segera ke dokter jika mengalami demam tinggi pasca transfusi, reaksi alergi (gatal, sesak, kemerahan), atau urine berwarna gelap setelah transfusi.
- Waspadai gejala kelebihan besi seperti kulit yang semakin menghitam, nyeri sendi, kelelahan ekstrem, atau gangguan irama jantung pada penderita yang rutin transfusi, dan segera laporkan ke dokter hematologi yang menangani.
- Penderita thalasemia yang mengalami nyeri dada, sesak napas mendadak, jantung berdebar keras, pingsan, atau penurunan kesadaran harus segera mendapat pertolongan darurat.
Cara Mengobati Thalasemia
🏠 Perawatan di Rumah
- Patuhi jadwal transfusi darah dan kontrol rutin ke dokter sesuai yang telah ditetapkan, karena keteraturan transfusi adalah kunci utama menjaga kadar hemoglobin dalam kisaran aman dan mencegah komplikasi organ.
- Konsumsi obat kelasi besi (iron chelation therapy) sesuai resep dokter secara disiplin dan teratur, karena kelasi besi sangat penting untuk mencegah penumpukan besi yang merusak jantung, hati, dan organ lainnya.
- Hindari konsumsi suplemen zat besi dan multivitamin yang mengandung zat besi tanpa anjuran dokter, karena penderita thalasemia umumnya sudah mengalami kelebihan besi, bukan kekurangan.
- Terapkan pola makan bergizi seimbang dengan asupan protein tinggi, asam folat (dari sayuran hijau, kacang-kacangan), dan kalsium untuk mendukung produksi sel darah merah dan kesehatan tulang — namun batasi makanan kaya zat besi berlebihan seperti hati dan suplemen besi.
- Jaga kebersihan diri dan lingkungan secara ketat, rajin mencuci tangan, dan hindari kontak dengan orang yang sedang sakit infeksi, karena penderita thalasemia (terutama yang sudah splenektomi) lebih rentan terhadap infeksi serius.
- Pastikan penderita mendapatkan semua vaksinasi yang direkomendasikan dokter, termasuk vaksin pneumokokus, meningokokus, dan influenza, terutama bagi yang telah menjalani splenektomi.
- Berikan dukungan psikologis dan emosional yang konsisten kepada penderita, terutama anak-anak dan remaja, karena hidup dengan penyakit kronis dapat berdampak signifikan pada kesehatan mental, kepercayaan diri, dan kualitas hidup mereka.
- Pantau pertumbuhan anak secara berkala dan catat perubahan gejala seperti perut yang semakin membesar, warna kulit, atau tingkat energi, untuk dilaporkan pada kunjungan berikutnya.
💊 Pengobatan Medis
- Transfusi sel darah merah (packed red cells/PRC) secara berkala adalah penatalaksanaan utama untuk thalasemia beta mayor, dengan tujuan mempertahankan kadar hemoglobin di atas 9–10 g/dL agar pertumbuhan dan perkembangan pasien dapat berlangsung normal.
- Terapi kelasi besi (iron chelation) menggunakan agen seperti desferoksamin (deferoxamine), deferasiroks, atau deferipron sangat penting untuk mengeluarkan kelebihan zat besi dari tubuh dan mencegah kerusakan organ vital akibat hemosiderosis.
- Transplantasi sel punca hematopoietik (bone marrow transplant/stem cell transplant) dari donor yang cocok (terutama saudara kandung yang sesuai HLA) saat ini merupakan satu-satunya terapi yang berpotensi menyembuhkan thalasemia secara permanen.
- Terapi gen (gene therapy) adalah pendekatan terkini yang tengah dikembangkan dan mulai mendapat persetujuan di beberapa negara, di mana gen normal dimasukkan ke dalam sel punca pasien untuk memperbaiki produksi hemoglobin secara permanen.
- Splenektomi (pengangkatan limpa secara bedah) dapat dipertimbangkan pada kasus hipersplenisme berat di mana limpa menghancurkan sel darah merah terlalu cepat dan kebutuhan transfusi menjadi sangat tinggi atau tidak efektif.
- Suplementasi asam folat diberikan untuk mendukung pembentukan sel darah merah baru, karena kebutuhan folat meningkat pada kondisi eritropoiesis yang aktif seperti pada thalasemia.
- Monitoring komplikasi dilakukan secara rutin melalui pemeriksaan ferritin serum, ekokardiografi jantung, MRI organ (untuk menilai deposit besi di jantung dan hati), densitometri tulang, dan evaluasi fungsi hati serta endokrin secara berkala.
- Penanganan harus dilakukan oleh tim multidisiplin yang terdiri dari dokter spesialis hematologi, anak, kardiologi, endokrinologi, dan gizi klinis, bekerja sama dalam program manajemen thalasemia komprehensif.
Pencegahan Thalasemia
- Lakukan skrining thalasemia sebelum menikah — pasangan muda sangat dianjurkan untuk menjalani pemeriksaan darah lengkap dan analisis hemoglobin untuk mengetahui apakah keduanya adalah pembawa (carrier) gen thalasemia.
- Jika kedua calon pasangan diketahui adalah pembawa thalasemia, konseling genetik pra-nikah bersama dokter spesialis sangat penting untuk memahami risiko dan pilihan yang tersedia sebelum merencanakan kehamilan.
- Diagnosis prenatal (sebelum lahir) tersedia untuk pasangan yang keduanya adalah carrier, melalui prosedur seperti chorionic villus sampling (CVS) pada trimester pertama atau amniosentesis pada trimester kedua, untuk mengetahui status genetik janin.
- Penyuluhan dan edukasi kepada masyarakat, terutama di daerah-daerah dengan prevalensi thalasemia tinggi, tentang pentingnya skrining sebelum menikah dan perencanaan kehamilan yang bertanggung jawab.
- Program skrining neonatal (bayi baru lahir) di rumah sakit memungkinkan deteksi dini thalasemia sehingga penatalaksanaan dapat dimulai sedini mungkin sebelum terjadi komplikasi.
- Dukung dan ikuti program skrining thalasemia yang diselenggarakan oleh pemerintah, puskesmas, atau rumah sakit di daerah Anda — di Indonesia, program ini semakin diperluas sebagai bagian dari upaya pencegahan nasional.
- Hindari pernikahan antar kerabat dekat (konsanguinitas) di komunitas dengan riwayat thalasemia yang tinggi, karena hal ini secara signifikan meningkatkan risiko anak mewarisi gen bermutasi dari kedua sisi keluarga.
Pertanyaan Umum tentang Thalasemia
Thalasemia adalah kelainan darah genetik yang menyebabkan produksi hemoglobin tidak normal sehingga memicu anemia kronis.
Thalasemia disebabkan oleh mutasi gen yang diwariskan dari orang tua ke anak (bersifat genetik).
Saat ini, satu-satunya potensi penyembuhan adalah transplantasi sumsum tulang, tetapi sebagian besar kasus hanya dapat dikontrol dengan pengobatan rutin.
Gejalanya meliputi lemas, pucat, pertumbuhan terhambat, pembesaran limpa, dan anemia.
Ya, terutama thalasemia mayor karena dapat menyebabkan komplikasi serius dan membutuhkan transfusi darah rutin.
Akademisi, peneliti, dan ahli farmasi dengan spesialisasi farmakologi bahan alam.
Disclaimer Medis: Informasi pada halaman ini bersifat edukatif dan tidak menggantikan saran, diagnosis, atau pengobatan dari tenaga medis profesional. Selalu konsultasikan kondisi kesehatan Anda dengan dokter atau apoteker yang kompeten.
Referensi
- WHO. Thalassaemia. World Health Organization. Tersedia di: https://www.who.int/news-room/fact-sheets/detail/thalassaemia
- Kemenkes RI. Pedoman Penatalaksanaan Thalasemia. Kementerian Kesehatan Republik Indonesia. 2018.
- POPTI (Perhimpunan Orangtua Penderita Thalassaemia Indonesia).
- Mayo Clinic. Thalassemia: Symptoms and causes. Tersedia di: https://www.mayoclinic.org/diseases-conditions/thalassemia
- MedlinePlus. Thalassemia. U.S. National Library of Medicine. Tersedia di: https://medlineplus.gov/thalassemia.html
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155-167.
- Cappellini MD, et al. Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT). 4th ed. Thalassaemia International Federation; 2021.
- PNPK (Panduan Nasional Praktik Klinis) Thalasemia. Kolegium Ilmu Kesehatan Anak Indonesia (IDAI). 2020.
- Muncie HL, Campbell JS. Alpha and Beta Thalassemia. Am Fam Physician. 2009;80(4):339-344.
- Musallam KM, et al. Non-transfusion-dependent thalassemias. Haematologica. 2013;98(6):833-844.